ATC code: C03DA05.
Pharmacology: Pharmacodynamics: Mechanism of Action: Finerenone is a nonsteroidal antagonist of the mineralocorticoid receptor (MR) that potently attenuates inflammation and fibrosis mediated by MR overactivation. The MR is expressed in the kidneys, heart and blood vessels where finerenone also counteracts sodium retention and hypertrophic processes. Finerenone has high selectivity for the MR due to its nonsteroidal structure and bulky binding mode. Finerenone has no relevant affinity for androgen, progesterone, estrogen and glucocorticoid receptors and therefore does not cause sex hormone-related adverse events (e.g., gynecomastia). Its binding to the MR leads to a specific receptor ligand complex that blocks recruitment of transcriptional coactivators implicated in the expression of pro-inflammatory and pro-fibrotic mediators.
Effects in healthy participants: Finerenone (multiple doses of 10 mg and 20 mg twice daily or 40 mg once daily over 10 days) had no consistent effect on natriuresis or urine potassium in healthy male participants. These regimens led to activation of the renin-angiotensin-aldosterone system (RAAS), i.e., reversible increases of plasma renin activity and serum aldosterone concentrations with baseline values reached again within 48 hours after the last dose.
However, following activation of the MR with the MR agonist fludrocortisone (0.5 mg), finerenone (single doses of 2.5, 5, 10, 20 mg PEG solution or 20 mg tablets) showed dose-dependent natriuretic effects in healthy male participants. Moreover, finerenone (at doses of 5 to 20 mg) significantly decreased urinary potassium excretion as compared to placebo.
Single or multiple doses of finerenone did not influence vital signs parameters in healthy participants.
Cardiac electrophysiology: In a thorough QT study in 57 healthy participants, there was no indication of a QT/QTc prolonging effect of finerenone after single doses of 20 mg (therapeutic) or 80 mg (supratherapeutic), indicating that finerenone has no effect on cardiac repolarization.
Clinical Trials: The FIDELIO-DKD study was a randomised, double-blind, placebo-controlled, multicentre Phase III study investigating the effect of KERENDIA compared to placebo on kidney and cardiovascular outcomes in adult patients with chronic kidney disease and type 2 diabetes. Patients were eligible based on evidence of persistent albuminuria (>30 mg/g to 5,000 mg/g), an eGFR of 25 to 75 mL/min/1.73 m
2, serum potassium ≤4.8 mmol/L at screening, and were required to be receiving standard of care, including a maximum tolerated labelled dose of an angiotensin-converting enzyme inhibitor (ACEi) or angiotensin receptor blocker (ARB).
The primary endpoint in the FIDELIO-DKD study was a composite of time to first occurrence of kidney failure (defined as chronic dialysis or kidney transplantation, or a sustained decrease in eGFR to <15 mL/min/1.73 m
2 over at least 4 weeks), a sustained decline in eGFR of 40% or more compared to baseline over at least 4 weeks, or renal death. The key secondary endpoint was a composite of time to first occurrence of cardiovascular (CV) death, non-fatal myocardial infarction (MI), non-fatal stroke or hospitalisation for heart failure.
The trial analysed 5,674 patients randomly assigned to receive either KERENDIA 10 mg or 20 mg once daily (N=2833), or placebo (N=2841), with a median follow-up duration of 2.6 years. After the end of study notification, vital status was obtained for 99.7% of patients. The trial population was 63% White, 25% Asian and 5% Black. The mean age at enrolment was 66 years and 70% of patients were male. At baseline, the mean eGFR was 44.3 mL/min/1.73 m
2, with 55% of patients having an eGFR <45 mL/min/1.73 m
2, median urine albumin-to-creatinine ratio (UACR) was 852 mg/g, and mean glycated haemoglobin A1c (HbA1c) was 7.7%, 46% had a history of atherosclerotic cardiovascular disease, 30% had history of coronary artery disease, 8% had a history of cardiac failure, and the mean blood pressure was 138/76 mmHg. The mean duration of type 2 diabetes at baseline was 16.6 years and a history of diabetic retinopathy and diabetic neuropathy was reported in 47% and 26% of patients at baseline, respectively. At baseline, almost all patients were on ACEi (34%) or ARB (66%), and 97% of patients used one or more antidiabetic medications (insulin [64%], biguanides [44%], glucagon-like peptide-1 [GLP-1] receptor agonists [7%], sodium-glucose cotransporter 2 [SGLT2] inhibitors [5%]). The other most frequent medications taken at baseline were statins (74%) and calcium channel blockers (63%).
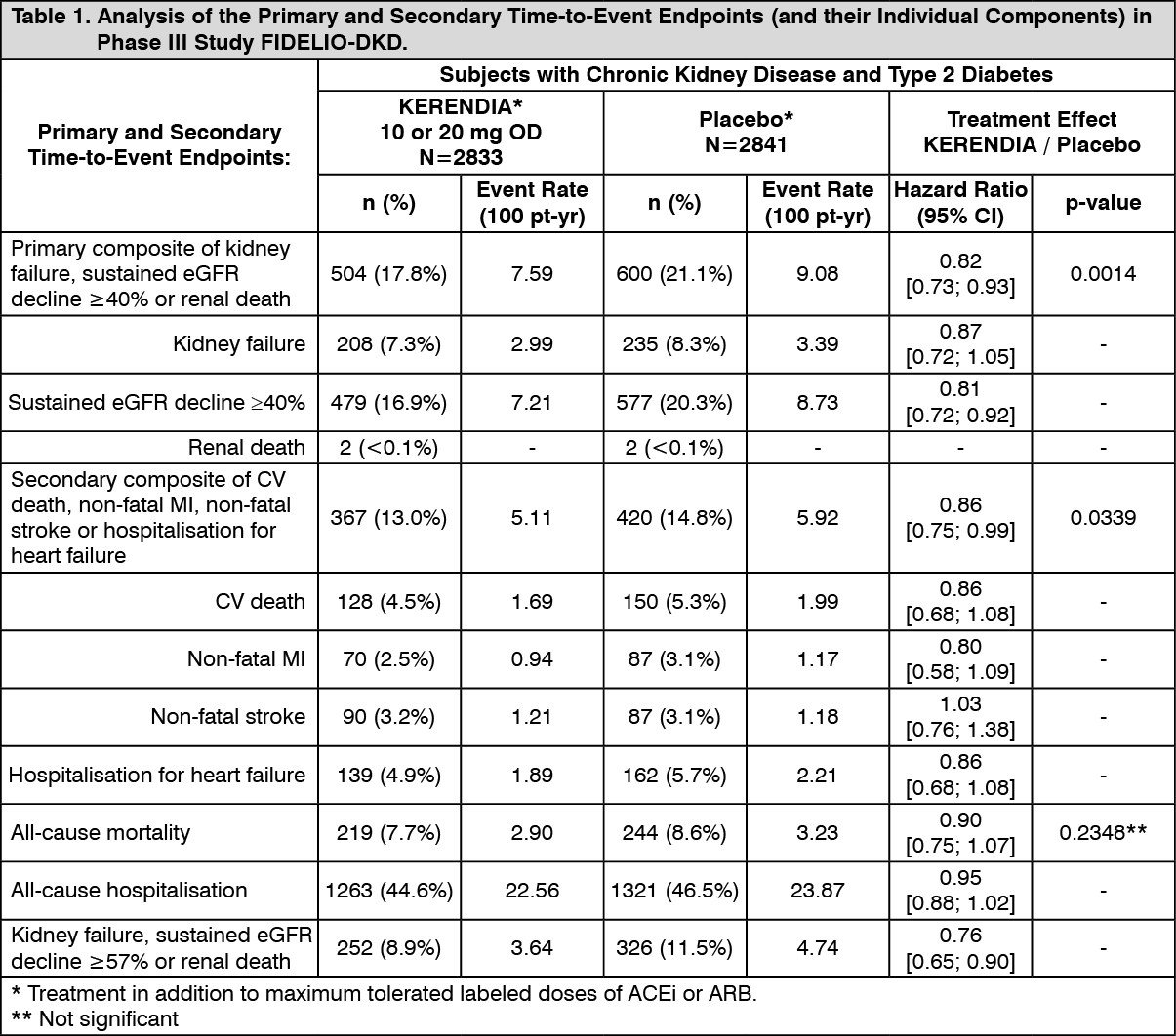
KERENDIA significantly reduced the risk of the primary composite endpoint compared to placebo in a time to event analysis using the Cox proportional hazards model and log rank test (HR 0.82, 95% CI 0.73-0.93, p=0.0014). See Figure 1/Table 1 as follows. The key secondary endpoint results (composite of CV death, non-fatal MI, non-fatal stroke, hospitalisation for heart failure) were favourable overall (HR (95% CI): 0.86 (0.75-0.99), p=0.0339), but with an indeterminate effect observed for the 'non-fatal stroke' component with a HR (95% CI): 1.027 (0.765-1.380). Prespecified secondary time-to-event endpoints are included in Table 1. For the secondary endpoint of change in UACR from baseline to month 4, a relative reduction of 31.2% was observed in the KERENDIA group compared to placebo. The treatment effect for the primary and key secondary endpoints was generally consistent across subgroups, including region, eGFR, UACR, systolic blood pressure (BP) and HbA1c at baseline.
In the FIDELIO-DKD study, hyperkalaemia events were reported in 18.3% of KERENDIA-treated patients compared with 9.0% of placebo-treated patients. Hospitalisation due to hyperkalaemia for the KERENDIA group was 1.4% versus 0.3% in the placebo group. Hyperkalaemia leading to permanent discontinuation in patients who received KERENDIA was 2.3% versus 0.9% in the placebo group.
In the FIDELIO-DKD study, glomerular filtration rate decreased events were reported in 6.3% of KERENDIA-treated patients compared with 4.7% of placebo-treated patients, and those leading to permanent discontinuation in patients receiving KERENDIA were 0.2% versus 0.3% in the placebo group. Patients on KERENDIA experienced an initial decrease in eGFR (mean 2 mL/min/1.73 m
2) that attenuated over time compared to placebo. This decrease has been shown to be reversible after treatment discontinuation. The initial decrease in eGFR was associated with long-term preservation of kidney function. (See Table 1 and Figures 1 and 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Pharmacokinetics:
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Pharmacokinetics: The concentration-effect relationship over time for UACR was characterised by a maximum effect model indicating saturation at high exposures. The model-predicted time to reach the full (99%) steady-state drug effect on UACR was 138 days. The pharmacokinetic (PK) half-life was 2-3 hours and PK steady state was achieved after 2 days, indicating timescale separation.
Absorption: Finerenone is almost completely absorbed after oral administration. Absorption is rapid with maximum plasma concentrations (C
max) appearing between 0.5 and 1.25 hours after tablet intake in the fasted state. The absolute bioavailability of finerenone is 43.5% due to first-pass metabolism in the gut-wall and liver. Finerenone is not a substrate of the efflux transporter P-gp
in vivo. Intake with high fat, high calorie food increased finerenone AUC by 21%, reduced C
max by 19% and prolonged the time to reach C
max to 2.5 hours. This is not clinically relevant. Therefore, finerenone can be taken with or without food (see Dosage & Administration).
Distribution: The volume of distribution at steady state (V
ss) of finerenone is 52.6 L. The human plasma protein binding of finerenone
in vitro is 91.7%, with serum albumin being the main binding protein.
Metabolism: Approximately 90% of finerenone metabolism is mediated by CYP3A4 and 10% by CYP2C8. Four major metabolites were found in plasma, resulting from oxidation of the dihydropyridine moiety to a pyridine (M1a, M1b), subsequent hydroxylation of a methyl group (M2a) and formation of a carboxyl function (M3a). All metabolites are pharmacologically inactive.
Excretion: The elimination of finerenone from plasma is rapid with an elimination half-life (t
1/2) of about 2 to 3 hours. Excretion of unchanged finerenone represents a minor route (<1% of dose in the urine due to glomerular filtration, <0.2% in the faeces). About 80% of the administered dose was excreted via urine and approximately 20% of the dose was excreted via faeces, almost exclusively in the form of metabolites. With a systemic blood clearance of about 25 L/h, finerenone can be classified as a low clearance drug.
Special populations: Patients with renal impairment: Mild renal impairment (CLCR 60 - <90 mL/min) did not affect finerenone AUC and C
max. Compared to subjects with normal renal function (CLCR ≥90 mL/min), the effect of moderate (CLCR 30 - <60 mL/min) or severe (CLCR <30 mL/min) renal impairment on AUC of finerenone was similar with increases by 34-36%. Moderate or severe renal impairment had no effect on C
max (see Dosage & Administration).
Due to the high plasma protein binding, finerenone is not expected to be dialyzable.
Patients with hepatic impairment: There was no change in finerenone exposure in cirrhotic subjects with mild hepatic impairment (Child Pugh A) (see Dosage & Administration).
In cirrhotic subjects with moderate hepatic impairment (Child Pugh B), finerenone mean AUC was increased by 38% and C
max was unchanged compared to healthy control subjects (see Dosage & Administration).
There are no data in patients with severe hepatic impairment (Child Pugh C) (see Dosage & Administration and Precautions).
Elderly patients: Of the 2827 patients who received KERENDIA in the FIDELIO-DKD study, 58% of patients were 65 years and older, and 15% were 75 years and older. No overall differences in safety or efficacy were observed between these patients and younger patients.
Elderly subjects (≥65 years of age) exhibited higher finerenone plasma concentrations than younger subjects (≤45 years of age), with mean AUC and C
max values being 34% and 51% higher in the elderly (see Dosage & Administration). Report No. R-13179 Population-pharmacokinetic analyses did not identify age as a covariate for finerenone AUC or C
max.
Body Weight: Population-pharmacokinetic analyses identified body weight as a covariate for finerenone C
max. The C
max of a subject with a body weight of 50 kg was estimated to be 43% to 51% higher compared to a subject of 100 kg. Dose adaptation based on body weight is not warranted (see Dosage & Administration).
Toxicology: Preclinical Safety Data: Genotoxicity: Finerenone was non-genotoxic in assays for mutagenicity in bacteria and for chromosomal aberrations
in vitro (in Chinese hamster V79 cells), and the mouse bone marrow micronucleus test.
Carcinogenicity: In 2-year carcinogenicity studies, finerenone did not increase tumour incidence in male or female rats at oral doses up to 20 and 10 mg/kg/day, or in female mice at oral doses up to 7.5 mg/kg/day (yielding exposure 19-28 times higher than in patients at the maximum recommended human dose of 20 mg/day, based on plasma AUC for unbound drug). In male mice, finerenone resulted in an increase in Leydig cell adenoma at 30 mg/kg/day, representing 26 times the AUC
unbound in humans. No carcinogenicity was evident with treatment at 10 mg/kg/day, representing 17 times the AUC
unbound in humans. Based on the known sensitivity of rodents to develop these tumours and the pharmacology-based mechanism at supratherapeutic doses as well as the margin of exposure, the increase in Leydig cell tumours observed in male mice is not considered to indicate a particular carcinogenic risk to patients treated with KERENDIA.



 Sign Out
Sign Out